Pompe's Disease Enzyme Deficiency
Pompe's Disease Enzyme Deficiency. Pompe disease is a rare neuromuscular genetic disease. Gaa is required for breaking down the glycogen, a complex carbohydrate which is converted by the body into glucose for energy.
 RNA splicing mutations and human disease Pompe disease. from www.slideshare.net
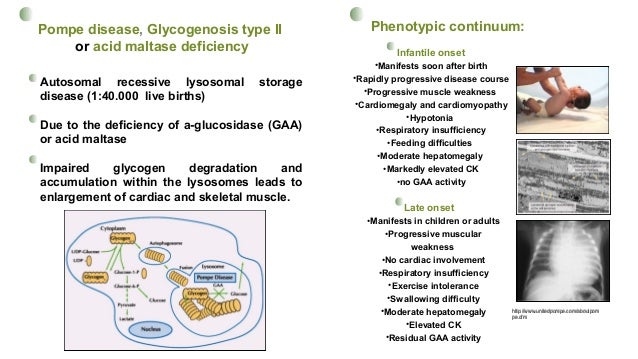
RNA splicing mutations and human disease Pompe disease. from www.slideshare.net1 it was the first recognized lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease. Pompe disease is an autosomal recessive disorder caused by a deficiency of the enzyme acid alpha glucosidase (gaa), which results in lysosomal accumulation of glycogen in. The severity of the disease and the age of onset are related to the degree of enzyme deficiency.

This enzyme is necessary to break down glycogen and to convert it into glucose. The enzymes affected normally catalyze reactions that ultimately convert glycogen compounds to monosaccharides, of which glucose is the predominant component.

Pompe disease is a genetic disorder in which complex sugar called glycogen builds up in the body's cells. After a decade of research, another therapeutic option,.

Pompe disease is a rare (estimated at 1 in every 40,000 births), inherited and often fatal disorder that disables the heart and skeletal muscles. They have significantly improved the outlook for people with acid maltase deficiency.

After a decade of research, another therapeutic option,. The enzymes affected normally catalyze reactions that ultimately convert glycogen compounds to monosaccharides, of which glucose is the predominant component.

Without sufficient gaa activity, massive amounts of glycogen can accumulate in the lysosome, causing distention of this organelle.1 1. 1 it was the first recognized lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease.
Pd can present with cardiac, skeletal muscle, and central nervous system manifestations, as a continuum of phenotypes among two main forms: Pompe disease (type ii glycogen storage disease) is an inherited enzyme defect that usually manifests in childhood.
1 it was the first recognized lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease. This enzyme deficiency causes excess amounts of glycogen to accumulate in the lysosomes,

This disease was originally referred to as pompe disease since joannes cassianus pompe (published in 1932) made the important observation of a massive accumulation of glycogen within the. Patients with pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure.
This disease was originally referred to as pompe disease since joannes cassianus pompe (published in 1932) made the important observation of a massive accumulation of glycogen within the. Mutations in the gaa gene cause pompe disease.

Pd can present with cardiac, skeletal muscle, and central nervous system manifestations, as a continuum of phenotypes among two main forms: This enzyme is necessary to break down glycogen and to convert it into glucose.

Pompe disease is a genetic disorder in which complex sugar called glycogen builds up in the body's cells. Pompe's disease is a rare, inherited, severe neuromuscular disease and often fatal disorder.
Pompe disease is a rare (estimated at 1 in every 40,000 births), inherited and often fatal disorder that disables the heart and skeletal muscles. Researchers have identified up to 300 different mutations in the gaa gene that cause the symptoms of pompe disease, which can vary widely in terms of age of onset and severity.
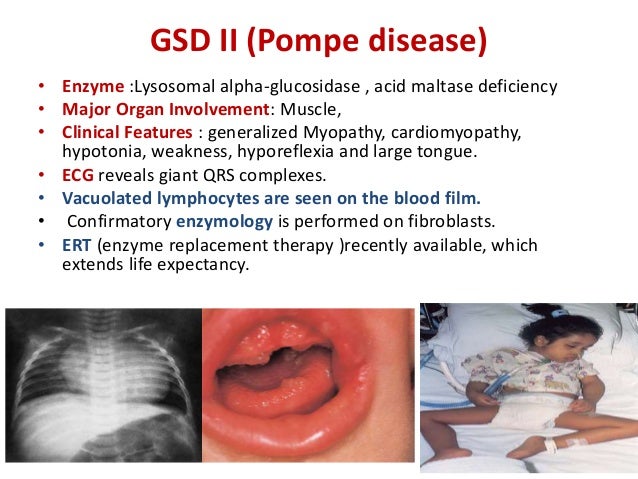
The severity of the disease and the age of onset are related to the degree of enzyme deficiency. They have significantly improved the outlook for people with acid maltase deficiency.

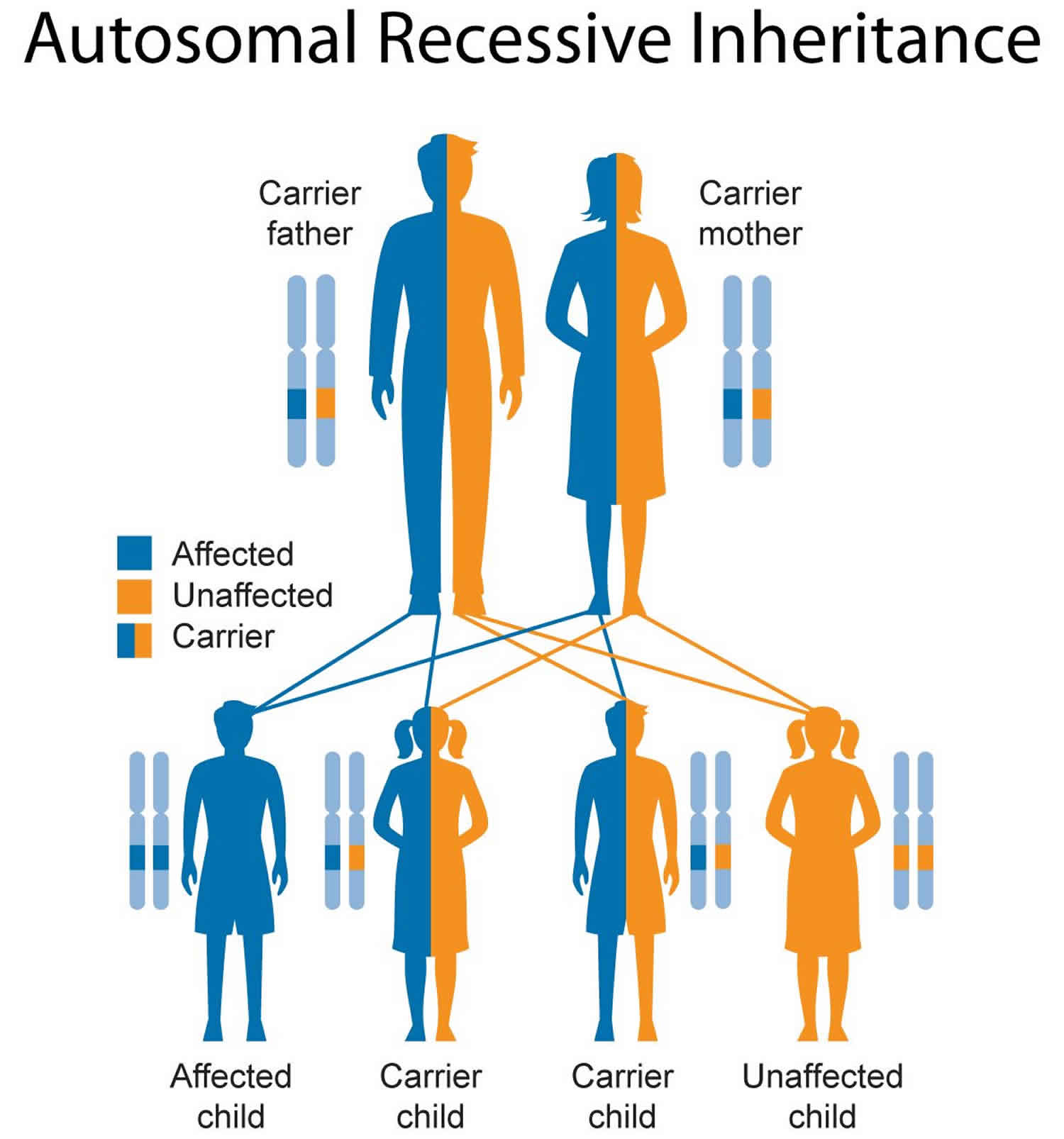
It is inherited as an autosomal recessive trait so that a couple presents a recurrent risk of 25% to have a child affected, at each pregnancy. Pd can present with cardiac, skeletal muscle, and central nervous system manifestations, as a continuum of phenotypes among two main forms:

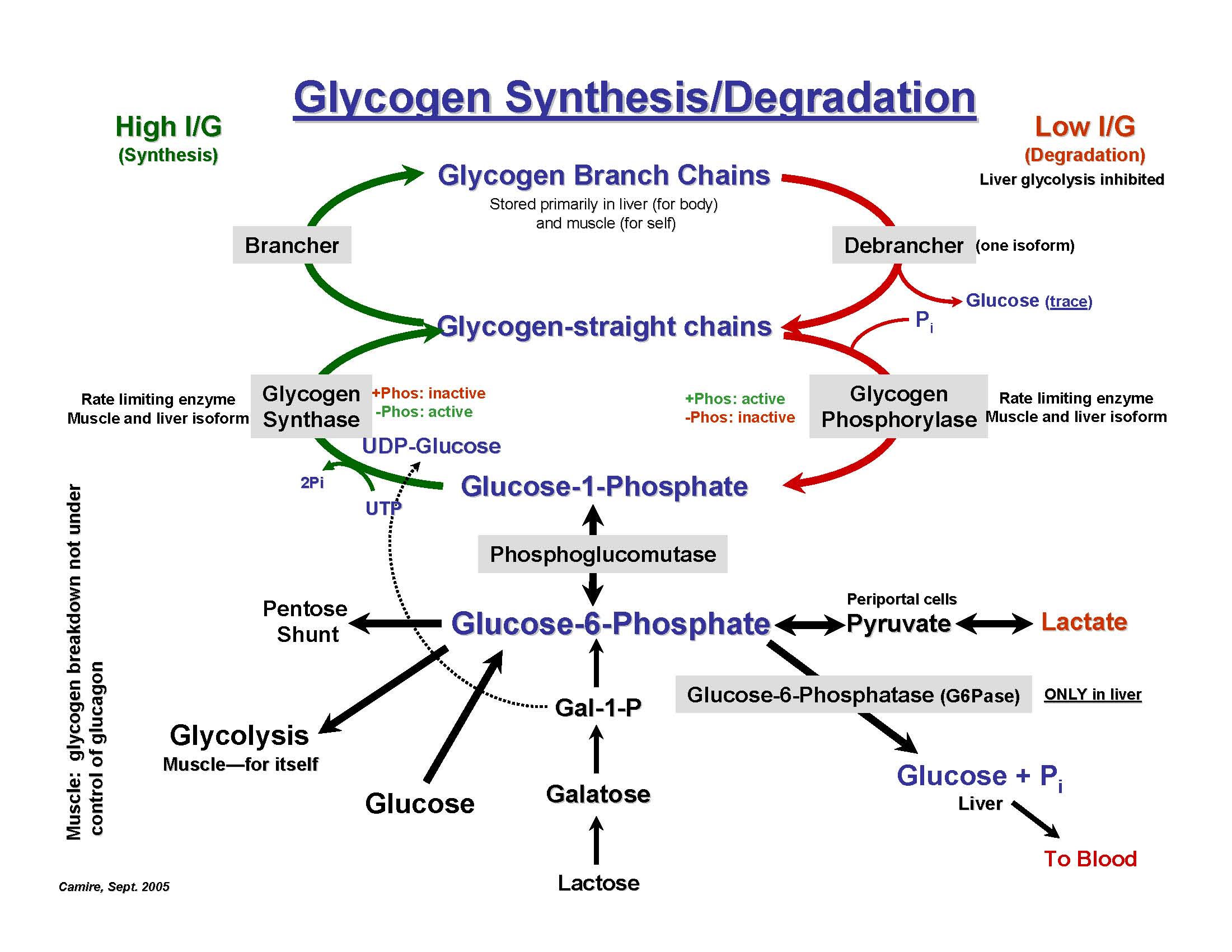
This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. This enzyme stops when there are four glucose units left before a branching point.
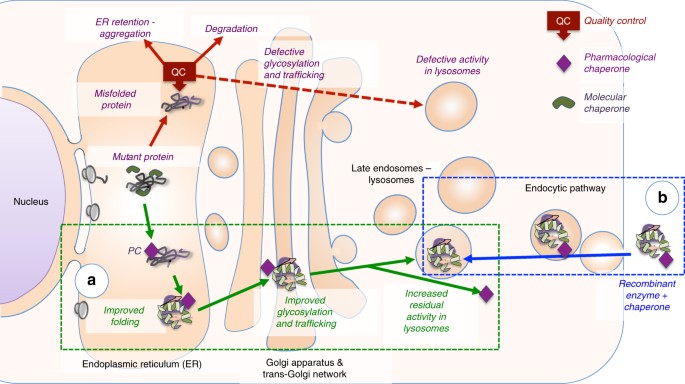
Pompe disease is a genetic disorder in which complex sugar called glycogen builds up in the body's cells. Acid maltase), which is necessary to break down glycogen, a substance that is a source of energy for the body.

This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. Acid maltase), which is necessary to break down glycogen, a substance that is a source of energy for the body.
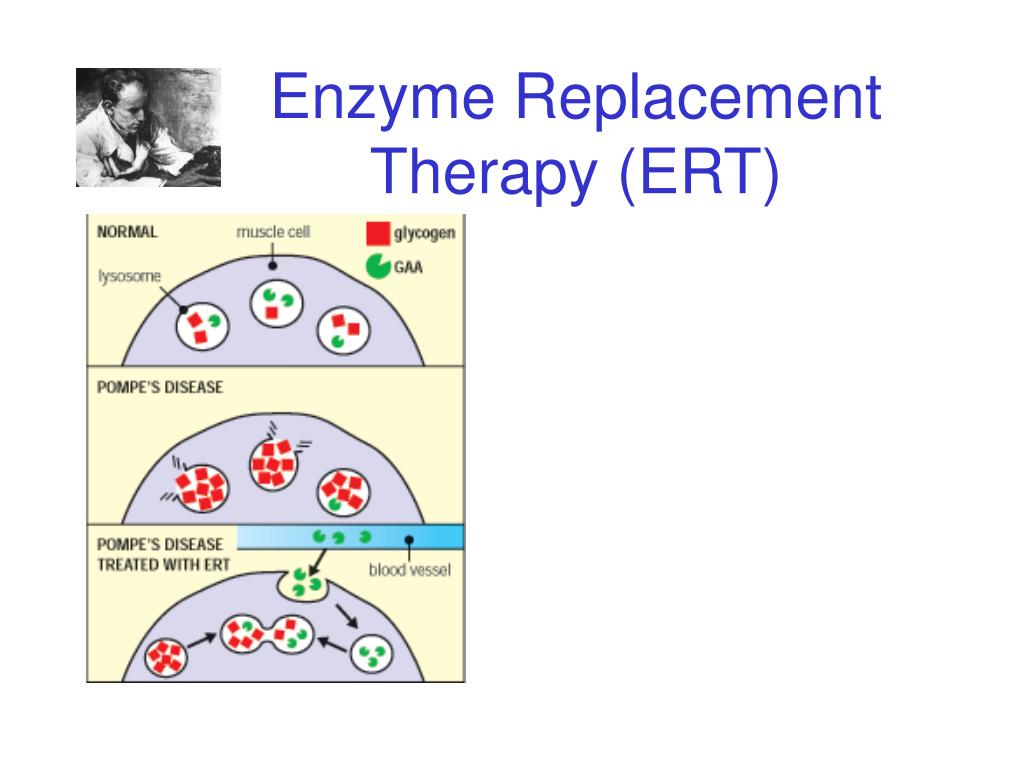
Both drugs substitute for the enzyme missing in pompe disease and may keep muscle cells from dying. Researchers have identified up to 300 different mutations in the gaa gene that cause the symptoms of pompe disease, which can vary widely in terms of age of onset and severity.
Without this enzyme, glycogen, a thick sticky substance, accumulates in the lysosomes (sacs within the muscle cells) and leads to severe muscle degradation. Pompe disease (type ii glycogen storage disease) is an inherited enzyme defect that usually manifests in childhood.

Acid maltase), which is necessary to break down glycogen, a substance that is a source of energy for the body. Gaa is required for breaking down the glycogen, a complex carbohydrate which is converted by the body into glucose for energy.
Glycogen Storage Disease Type 2 (Gsd2) Is An Autosomal Recessive Disorder That Is More Commonly Known As Pompe Disease Or Acid Maltase Deficiency (Amd).This disease was originally referred to as pompe disease since joannes cassianus pompe (published in 1932) made the important observation of a massive accumulation of glycogen within the. This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. They have significantly improved the outlook for people with acid maltase deficiency.
Pompe Disease Is A Rare (Estimated At 1 In Every 40,000 Births), Inherited And Often Fatal Disorder That Disables The Heart And Skeletal Muscles.Pompe's disease is a rare, inherited, severe neuromuscular disease and often fatal disorder. Acid maltase), which is necessary to break down glycogen, a substance that is a source of energy for the body. Pd can present with cardiac, skeletal muscle, and central nervous system manifestations, as a continuum of phenotypes among two main forms:
The Severity Of The Disease And The Age Of Onset Are Related To The Degree Of Enzyme Deficiency.1 it was the first recognized lysosomal storage disease and is the only glycogen storage disease that is also a lysosomal storage disease. Mutations in the gaa gene cause pompe disease. After a decade of research, another therapeutic option,.
Without Sufficient Gaa Activity, Massive Amounts Of Glycogen Can Accumulate In The Lysosome, Causing Distention Of This Organelle.1 1.Both drugs substitute for the enzyme missing in pompe disease and may keep muscle cells from dying. Pompe disease is an autosomal recessive disorder caused by a deficiency of the enzyme acid alpha glucosidase (gaa), which results in lysosomal accumulation of glycogen in. The disease results from the deficiency of an enzyme called acid alfa glucosidase (gaa), which breaks downs complex sugars in the body.
Researchers Have Identified Up To 300 Different Mutations In The Gaa Gene That Cause The Symptoms Of Pompe Disease, Which Can Vary Widely In Terms Of Age Of Onset And Severity.Pompe disease is a genetic disorder in which complex sugar called glycogen builds up in the body's cells. This enzyme stops when there are four glucose units left before a branching point. Pompe disease (type ii glycogen storage disease) is an inherited enzyme defect that usually manifests in childhood.
Belum ada Komentar untuk "Pompe's Disease Enzyme Deficiency"
Posting Komentar